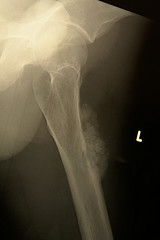
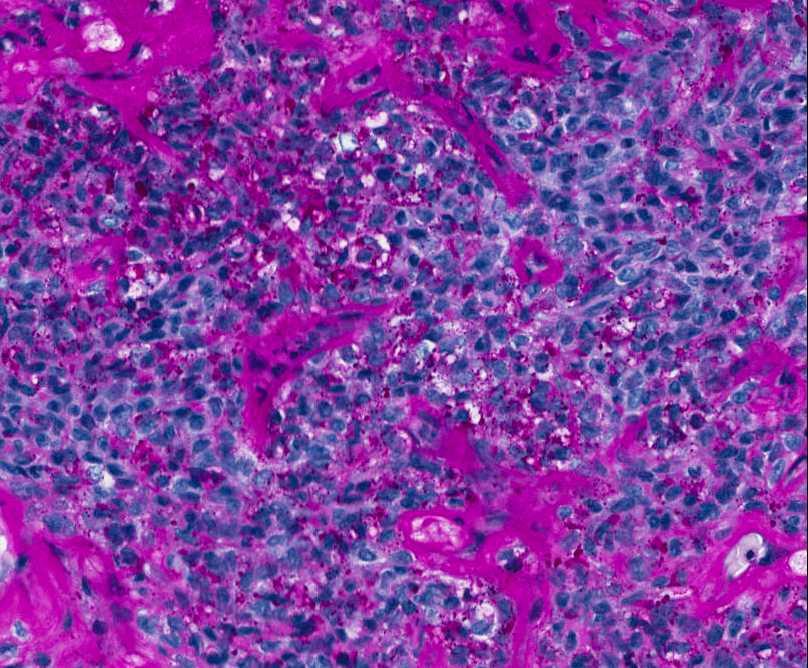
CONVENTIONAL OSTEOSARCOMA
Definition:
Primary intramedullary high grade malignant sarcoma in which neoplastic cells produce osteoid.
Epidemiology:
Most common primary non-hematopoietic malignancy of bone.
Its incidence in USA is 4-5 per milion individuals with 1000-1500 new cases diagnosed annually which
accounts for approximatelly 20% of all primary malignant
cell tumors.
It most frequently occurs in second decade of life with 60% of tumors occuring in patients younger than 25
years.
Although 30% occuring in patients over 40 years of age, the predisposing condition should always be considered
in older patients (e.g. Paget's disease, of bone, post
radiation sarcoma).
Male to female ratio 3:2. Osteosarcoma in children 5 years and younger is very uncommon, and they account for less than 2% of
osteosarcomas in the pediatric population.
Sites of involvement:
Conventional osteosarcoma shows a profound propensity for involvement of the long bones of the
appendicular skeleton; in particular , the distal femur,
proximal tibia, and proximal humerus.
Within the long bones the tumor is mostly centered in metaphysis (90%) followed by diaphysis (9%), and
rarely in epiphysis.
Although the long bones remain the most frequent site of involvement, the non-long bone (i.e, jaws, pelvis,
spine, and skull) involvement tends to increase with
age.
Clinical features:
Classic presentation of conventional osteosarcoma typically is progressively enlarging, painfull mass.
Deep seated and boring in nature, the pain is frequently noted months prior to diagnosis, and usually increases
in intensity over time, eventually becoming unbearable.
Skin overlying the tumor may be warm, eyrthematous, edematous, with prominent engorged veins. Large
tumors may restrict range of motion,decrease
musculoskeletal function,produce joint effusion and, in
advances cases,result in weight loss and cachexia.
A sudden fracture through destructive mass may be found.
Imaging:
Osteosarcoma has extremely variable radiographic appearances;
Conventional tumors present as a large, destructive, poorly defined, mixed lytic and blastic
masses accompanied by cortical destruction and
tumor extension into soft tissue.
Tumor/periosteal interaction may lead to variety of manifestations secondary to periosteal elevation (e.g.
Codman triangle) and periosteal reactive bone
formation.
Occasionally the tumor may demonstrate perpendicular or radiating striations called "sunburst".
CT and MRI may be helpfull in delineating the extent
of the tumor preoperatively.
Gross:
Osteosarcoma is often a large (over 5 cm), metaphyseally centered, fleshy or hard tumor which
may contain cartilage.
It frequently transgresses the cortex and is associated with a soft tissue mass.
Histopathology:
Presents as highly anaplastic, pleomorphic tumor in which the tumor cells may be: epithelioid,
plasmacytoid, fusiform, ovoid, small round cells, clear
cells, mono- or multinucleated giant cells, or spindle
cells producing osteoid.
Conventional osteosarcoma can also produce varying amounts of cartilage and/or fibrous tissue.
Many investigators further subdivide conventional osteosarcoma in terms of predominant matrix.
This divides conventional sarcoma into three subtypes:
osteoblastic (50%), chondroblastic (25%), and fibroblastic (25%) osteosarcoma. Osteoblastic osteosarcoma:
Bone and/or osteoid are the predominant matrix. The extremes of matrix production are thin arborising osteoid to dense, compact osteoid and bone
(sclerotic).
Chondroblastic osteosarcoma:
Chondroid matrix is predominant. It tends to be high grade hyaline cartilage, which is intimately associated, and randomly mixed, with
non-chondroid elements.
Fibroblastic osteosarcoma:
A high grade spindle-cell malignancy with sometimes minimal amount of osseous matrix with
or without cartilage.
In general, the overall histological appearance is similar to fibrosarcoma or pleomorphic
undifferentiated sarcoma (malignant fibrous
histiocytoma).
Genetics:
Loss of heterozygosity of chromosome arm 3q, 13q, 17p, and 18q most frequent.
Amplification at 1q21-23 and 17p are frequent findings in conventional osteosarcoma.
Prognosis:
Untreated, conventional osteosarcoma is fatal. Aggresive local growth and rapid hematogenous systemic dissemination are bad prognostic features.
Although metastases may affect many sites, pulmonary metastases are the most frequent site of
clinically significant systemic disease.
In children the treatment of conventional osteosarcoma is tailored to the location, size and
stage of the tumor.
Eradication of the primary tumor and the elimination of any matastases is the goal of therapy.
Limb salvage resection is the convention for appendicular tumors, and surgical excision in
combination with radiation is employed for tumors
that are not resectable entirely with negative
margins.
Adjuvant chemotherapy is usually employed in the preoperative setting and continued after surgical
resection.
Survival is directly related to response to pre-operative therapy. In those patients whose
tumors have >90% tumor necrosis, long-term
survival is generally 80-90%.
In patients with < 90% response to therapy the survival is poor, usually <15%.
MALIGNANT BONE TUMORS
BONE PRODUCING TUMORS
SMALL BLUE CELL TUMORS
REFERENCES:
WHO Pathology and Genetics of Tumors of Soft Tissue and Bone, Lyon: IARC Press, 2002
Dorfman H. D., Bone Tumors, New York: Mosby, 1998
Potter's, Pathology of the fetus, infant and child, Mosby/Elsevier 2007
Weiss W.S., Soft Tissue Tumors, Mosby/Elsevier 2008
TELANGIECTATIC OSTEOSARCOMA
Definition:
• spaces filled with blood with or without septa.
Epidemiology:
• osteosarcoma.
• was described in younger patients.
• Sites of involvement:
• bones.
• anatomic site, followed by the upper tibia and proximal
humerus or proximal femur.
Clinical findings:
• • fracture (1/4 of the cases),because of massive bone
destruction.
Imaging:
• surrounding bony sclerosis.
• • extend into the epiphysis.
• cortex.
• skin are frequent.
• intensity, and T2-weighted image shows high signal
intensity with several cystic foci and fluid-fluid level with an
extraskeletal extension of the tumor, similar to aneurysmal
bone cyst.
Gross:
• space.
• clot which is described as "a bag of blood".
• Histopathology:
• thin septa simulating aneurysmal bone cyst.
• spaces.
• benign-looking giant cells without endothelial cells.
• cells.
• mitotic activity including atypical mitoses.
• osteoid is observed in minimal amount.
• multinucleated giant cells, which may lead to a mistaken
diagnosis of benign or even malignant giant cell tumor.
Genetics:
• Prognosis:
• • SMALL CELL OSTEOSARCOMA
Definition:
• of steoid production.
Epidemiology:
• osteosarcomas.
• in the second decade.
• Sites of involvement:
• Clinical findings:
• Imaging:
• • radiodense areas.
• soft tissue tumor extension.
Histopathology:
• associated with osteoid production.
• small to medium; the smaller one are comparable to those
of Ewing sarcoma or lymphoma.
• round to oval and the chromatin may be fine to coarse.
• • to spindle, have a granular chromatin, inconspicuous
nucleoli and scanty amounts of cytoplasm.
• • especially from the peripheral surface of the tumor, because
may not always show osteoid and may resemble Ewing
sarcoma.
• diagnostic tool, which is negative in small cell
osteosarcoma.
Immunohistochemistry:
• Genetics:
• Prognosis:
•
LOW GRADE CENTRAL OSTEOSARCOMA
Definition:
• cavity of bone.
Epidemiology:
• 1-2% of all osteosarcomas.
• • Sites of invovement:
• located in the long bones with predilection for the distal
femur and proximal tibia.
• Clinical features:
• • years.
Imaging:
• • bone when the epiphyseal plate is closed.
• show intermediate or well defined margins suggestive of
indolent or benign lesion.
• reflects indolent nature of this tumor.
• feature to support malignant nature of the tumor.
• some degree of cortical destruction with or without soft
tissue extension.
Gross:
• grey-white tumor with a firm and gritty texture arising from
within the medullary cavity.
Histopathology:
• moderately cellular fibroblastic stroma with variable amounts
of osteoid production.
• interlacing bundles thet permeate surrounding pre-existing
bony trabeculae and bone marrow similar to that of
desmoplastic fibroma.
• • • • • and curved trabeculae simulating the appearance of woven
bone in fibrous dysplasia ( no atypia seen in spindle cells of
fibrous dysplasia).
• seen.
• reported in up to 36% of cases.
Genetics:
• 12p, and 6p21.
Prognosis:
• indolent fashion conventional osteosarcoma.
• • the potential for metastases.
PAROSTEAL OSTEOSARCOMA
Definition:
• arises on the surface of bone.
Epidemiology:
• type osteosarcoma of the surface of bone.
• • • 3rd decade of life.
Sites of involvement:
• • Clinical findings:
• symptom.
• Imaging:
• base.
• • involvement.
Gross:
• attached to the undrelying cortex.
• • covering the surface and thus suggesting a diagnosis of
osteochondroma.
Histopathology:
• trabeculae in spindle cell stroma.
• with difficulty, atypical forms are not present.
• simulate normal bone.
• • differentiation. This may be in the form of hypercellular
nodules of cartilage within the tumor or as a cap on the
surface.
• and the cells may show mild atypia and lack columnar
arrangemet seen in osteochondroma.
• osteochondromas, there is spindle cell proliferattion
between bony trabeculae.
• (dedifferentiation).
Prognosis:
•
PERIOSTEAL OSTEOSARCOMA
Definition:
• osteosarcoma arising on the surface of the bone.
Epidemiology:
• • surface high grade osteosarcoma, but about 1/3 as common
as parosteal osteosarcoma.
• • Sites of involvement:
• the tibia and femmur most commonly affected.
Clinical features:
• finding with pain and tenderness later developing in the
affected area.
Imaging:
• calcified spiculations that are disposed perpendicular to the
cortex and give overall sunburst appearance.
• surface, where the tumor has a relatively well demarcated
margin.
• production of ossified matrix.
• • integrity of the cortex, and soft tissue extension.
Gross:
• the bone or the entire circumference.
Histopathology:
• differentiated chondroblastic osteosarcoma.
• is made up of relatively mature bone.
• intermediate grade osteosarcoma are invariably present.
• cytological atypia and matrix may be myxoid.
• fascicles of spindle cells.
Prognosis:
• have tendency to recur and metastasize.
• • • HIGH GRADE SURFACE OSTEOSARCOMA
Definition:
• surface of the bone.
Epidemiology:
• • • Sites of involvement:
• the humerus and tibia.
Clinical features:
• of the tumor.
Imaging:
• • periosteal new bone is commonly present at the periphery of
the tumor.
• involvement, but the tumor is most comonly relateively well
circumscribed at its soft tissue margin.
Gross:
• erodes the underlying cortical bone.
Histopathology:
• • fibroblastic differentiation may predominate.
• and lace-like osteoid as seen in conventional osteosarcoma.
Prognosis:
• response to chemotherapy.
SECONDARY OSTEOSARCOMA
Definition:
• preexisting abnormalities the most common
being postradiation therapy chnges, Paget disease, and rarely
various other disorders.
POSTRADIATION OSTEOSARCOMA
Epidemiology:
• radiation-induced sarcomas.
• irradiated bone is 0.03-0.8%.
• chemotherapy are at greatest risk.
• as children survive treatment of their malignant disease.
Sites of involvement:
• pelvis and the shoulder region.
Clinical findings:
• path of radiation beam.
• years), and inversely related to the radiation dosage.
• • h/o of synovial sarcoma radiation therapy in young children.
Imaging:
• • coarsening and lytic areas in cortex).
Histopathology:
• osteosarcma).
Prognosis:
• lesions, 27.3% for patients with axial lesions.
PAGET OSTEOSARCOMA
• osteosarcomas represent 50-60% of Paget sarcomas.
• (M:F 2:1), with an overall median age of 64 years.
• population.
OSTEOSARCOMA ASSOCIATED WITH FIBROUS DYSPLASIA
• called McCune-Allbright syndrome and polyostotic fibrous
dysplasia = bone polyostotic fibrous dysplasia + skin
pigmentation and precosious puberty (other endocrine
abnormalities)
EWING SARCOMA/ PRIMITIVE NEUROECTODERMAL TUMOR (PNET)
Definition:
• (belong to group of pediatric small blue cell tumors) that show
varying degrees of neuroectodermal differentiation.
• neuroectodermal differentiation, and PNET has been described
for tumors with neuroectodermal differentiation.
Epidemiology:
• • • • during the second decade of life.
Sites of involvement:
• long bones.
• Clinical features:
• clinical symptoms.
• are often seen.
Imaging:
• flat bone is the most common feature.
• with "onion-skin" like multilayered periosteal reaction is
characteristic.
• thickened.
• • be seen.
Gross:
• and hemorrhagic.
Histopathology:
• round nuclei containing fine chromatin, scanty clear or
eosinophilic cytoplasm, and indistinct cytoplasmic
membranes, whereas in others, the tumor cells are larger, have
prominent nucleoli, and irregular contours.
• glycogen ( diastase sensitive).
• • distribution.
Immunophemotype:
• PAS+ diastase sensitive.
• Genetics:
• • • Prognosis:
• • location and the size of the tumor.
• in the pelvis, and when they are large tend to do poorly.
LYMPHOMA OF BONE
Definition:
• lymphoid cells, producing a intra-medullary tumor mass.
Epidemiology:
• for approximately 7% of all bone malignancies.
• lymphoma.
• involve adults, especially older patients, although there were
cases of young childrens causing difficult diagnostic
differentiation from Ewing sarcoma.
Sites of involvement:
• • • the small bones of the hand and feet.
• • • • systemic symptoms like fever or night sweats.
• 1) an single skeletal site, with or without regional lymph node
involvement;
2) multiple bones are involved, but there is no visceral or lymph
node involvement;
3) patients present with a bone tumor but work up shows
involvement of other visceral sites or multiple lymph node
sites;
4)patient has a known lymphoma and the bone biopsy is done to
r/o involvement of bone;
Groups 1 and 2 are considered primary lymphoma of bone.
Imaging:
• • to see destruction of more than half of the bone.
• from normal bone.
• sclerotic or entirely lytic.
• • mass.
• • obvious on plain roentgenograms.
• Gross:
• • Histopathology:
• pattern.
• marrow, although in most of the cases does not present as
destructive bone tumor.
• tumefactive process.
• cell type.
• • or irregular, even Pagetoid. 92% of primary non-Hodgkin
lymphoma of bone was found to be of the large B cell type and
only 3% diffuse follicle centre cell, #% anaplastic large cell and
2% immunocytoma.
• the calls tend to get crushed.
• malignant lymphoma should be suspected.
• manifestation of a wide spread disease and produce a tumor
mass but primary manifestations are rare.
Prognosis:
• stage of disease.
• worse progression-free period.
• than the centroblastic mono/polymorphic subtype or the
centroblastic multilobulated subtype.
Please email me your comments about the site.
The information provided on this site is designed to support, not replace, the relationship that exists between a patient/site visitor and his/her existing physician. Banners on the Home Page and at the top of other pages are advertisements. Our editorial content is free of any commercial influence. There are no medical or personal patient informations included.
The site is supported by advertisement. I value our advertisers, who make this free website possible.
Pedorthpath.com is concerned with online privacy for all visitors and advertisers at our site. Any information sent to us by email or otherwise, except for information intended to be posted at the website, is treated as confidential, and will not be disclosed to any third party without the express permission of the visitor or advertiser.
Last modification date: 03/15/2010.
Site designed and maintained by Dariusz Borys, M.D
CHONDROSARCOMA
Definition:
• • Epidemiology:
• bone tumours.
• myeloma and osteosarcoma.
• • incidence is in the fifth to seventh decades of life.
• • usually present as high grade.
Sites of involvement:
• ilium is the most frequently involved bone) followed by the
proximal femur, proximal humerus, distal femur and ribs.
• primary CHS (1% of all CHS).
• Clinical findings:
• presenting symptoms.
• years).
Imaging:
• diaphysis were the produce fusiform expansion with cortical
thickening of the bone.
• bone.
• distributed punctate or ring-like opacities(mineralization).
• • absent.
• establishing the presence of soft tissue extension.
• Gross:
• white color corresponding to the presence of hyaline cartilage.
• • cystic areas.
• tissue may be present expecially in CHS of the flat bones
pelvis, scapula and sternum).
Histopathology:
• matrix production.
• • trabeculae.
• • contain enlarged, hyperchromatic nuclei. Extent of atypia is
usually mild to moderate.
• • characteristic of CHS that can be used to separate it from
enchondroma.
• feature of chondrosarcomas.
• particularly in high grade lesions.
• Increased cellularity, binucleated cells, hyperchromasia and
myxoid change may all be present in enchondroma in this
location.
• small bones is permeationthrough the cortex into soft tissue
and a permeative pattern in the cancellous bone.
• • hyperchromatic plumpnuclei of uniform size. Occasionally
binucleated cells are present. The cytology is very similar to
enchondroma.
• degree of nuclear atypia, hyperchromasia and nuclear size.
• than grade 2.
Prognosis:
• combined group of patients with grade 2 and 3 have a five-year
survival of 53%.
• the degree of malignancy.
CARTILAGE PRODUCING TUMORS